
In the current version of LLPSDB, natural and designed proteins account for 85% and 15% of overall entries, respectively. The statistics here focuses on natural proteins with non-redundant sequences. Given that intrinsically disordered regions (IDRs) have been suggested to play critical roles in LLPS, the subsets that only one component of IDR-contained protein undergoing LLPS with or without nucleic acid (labeled as “Protein(1)+Nucleic acid” and “Protein(1)” in the plots below, respectively) were used in the statistical analysis. Meanwhile, because low complexity regions (LCRs), in which specific amino acids are overrepresented compared to the amino acid proportions in proteome, have been suggested to be likely important players in mediating protein LLPS, therefore, the residues within LCRs in the selected entries were also analyzed. IDRs (no less than 15 amino acids) were identified by searching MobiDB (Piovesan et al., 2018) or by PONDR VL3-BA algorithm (Obradovic et al., 2003) if the data are not available in MobiDB. LCRs were predicted through the SEG algorithm (Wootton, 1994) with default parameter.
The statistical plots below include the amino acid components of the whole sequence, the LCRs, as well as the folded and disordered regions of the proteins in the entries within the selected subsets. The sequence length distributions of the whole sequences and LCRs are also presented. The corresponding statistical results of the human proteome (refseq30 from NCBI) are used as a control (all residues are used except in the analysis of folded and disordered regions). Additionally, the distribution of parameter SCD (sequence charge decoration) which characterizes the charged residues pattern in a protein sequence (Sawle and Ghosh, 2015) is ploted, using the whole sequences in the subsets.
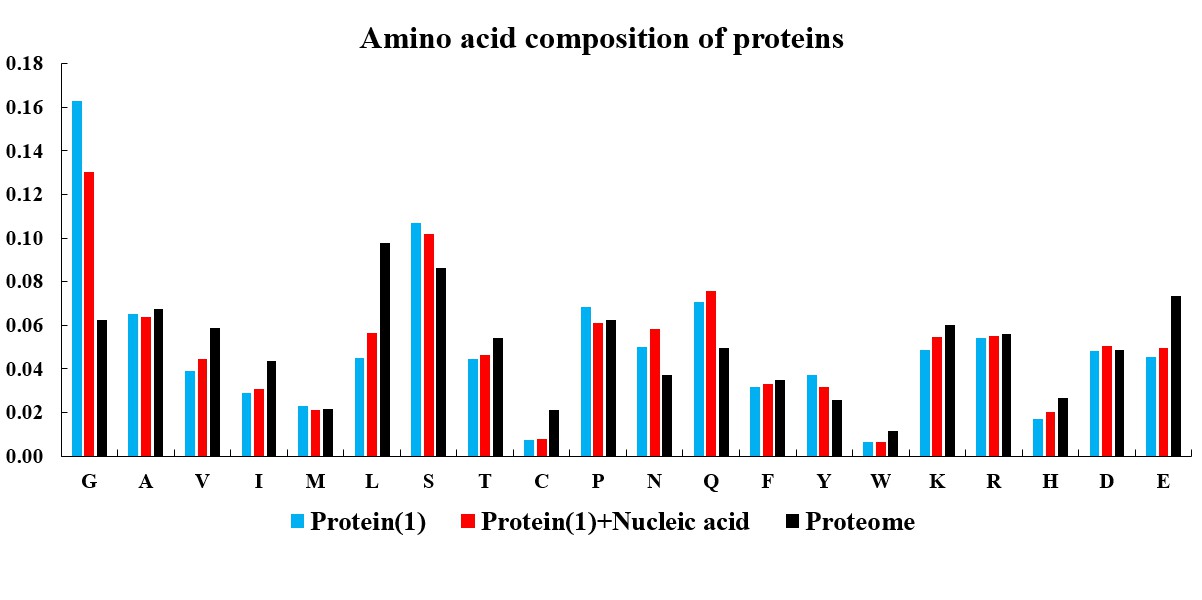
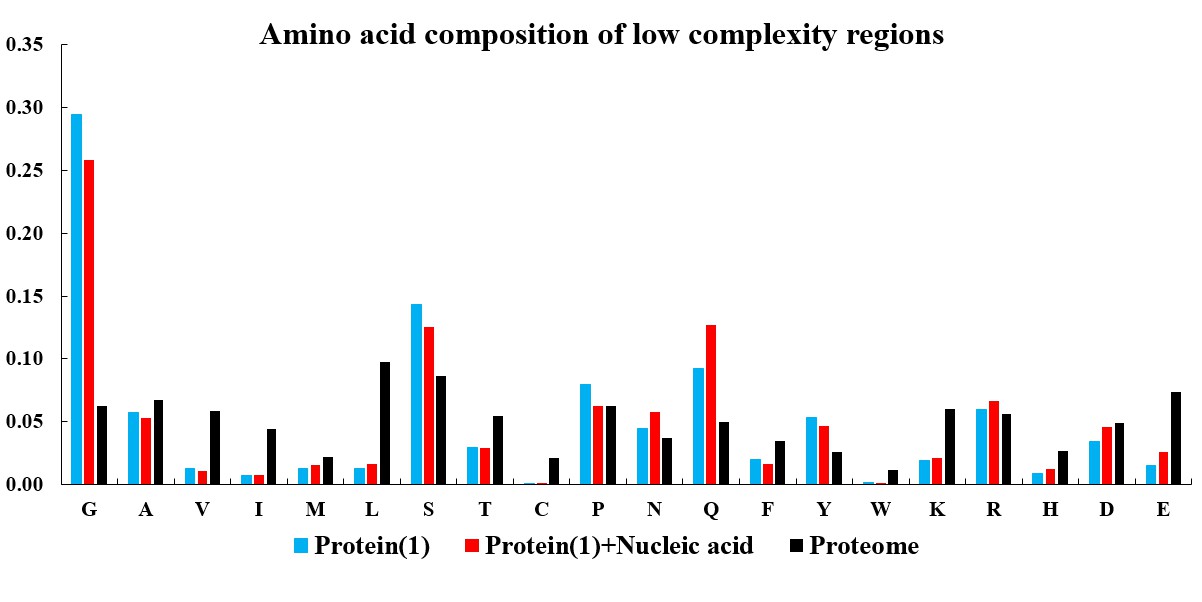
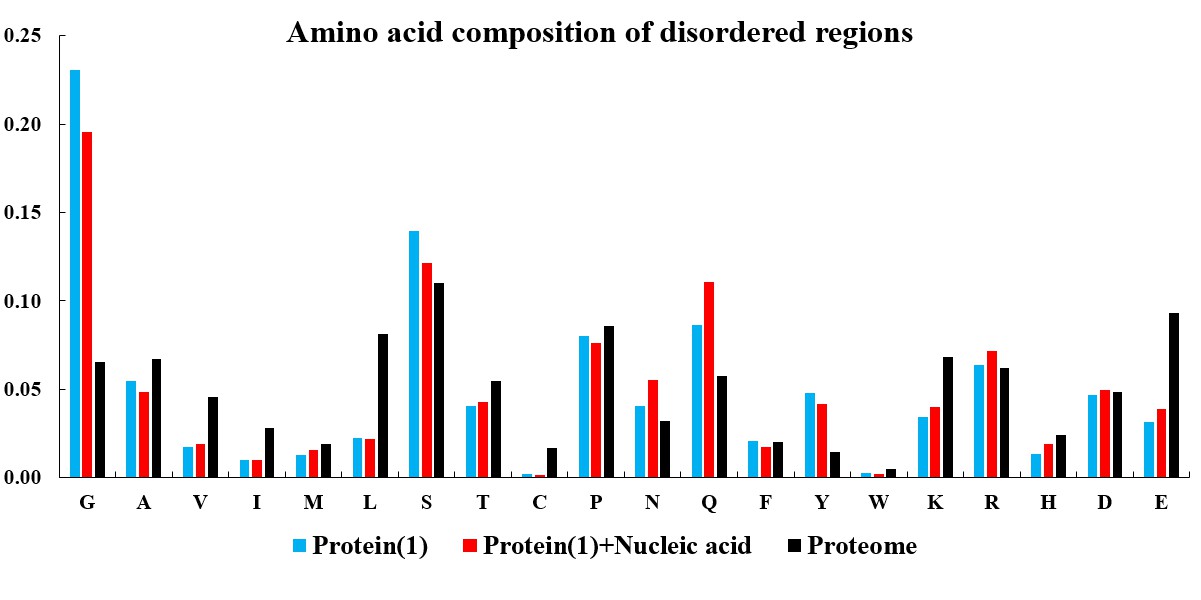
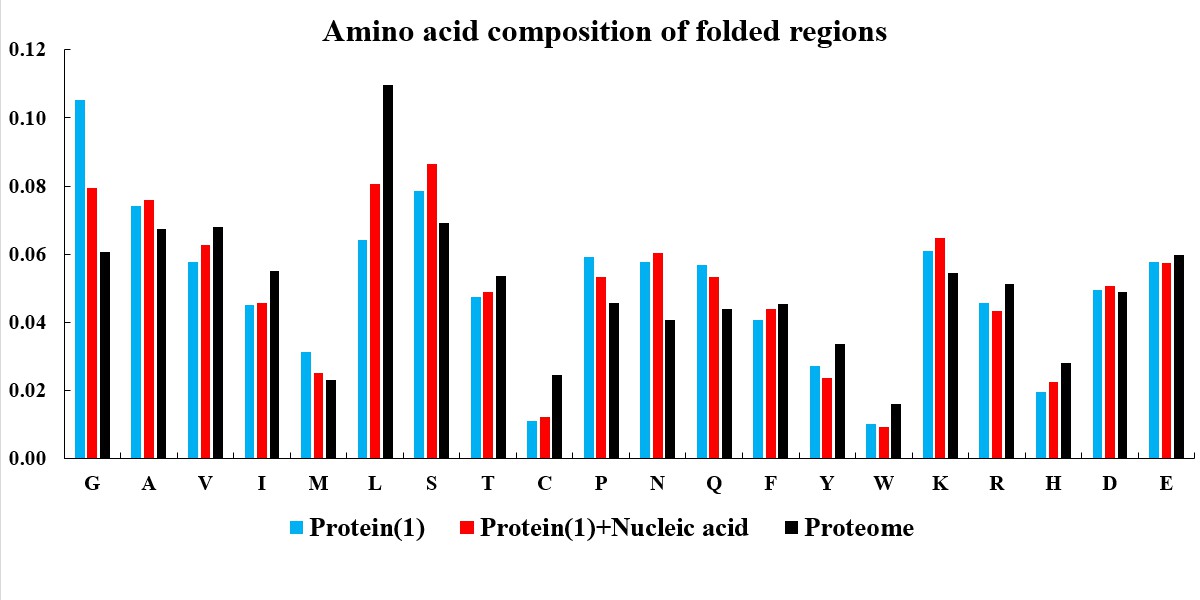
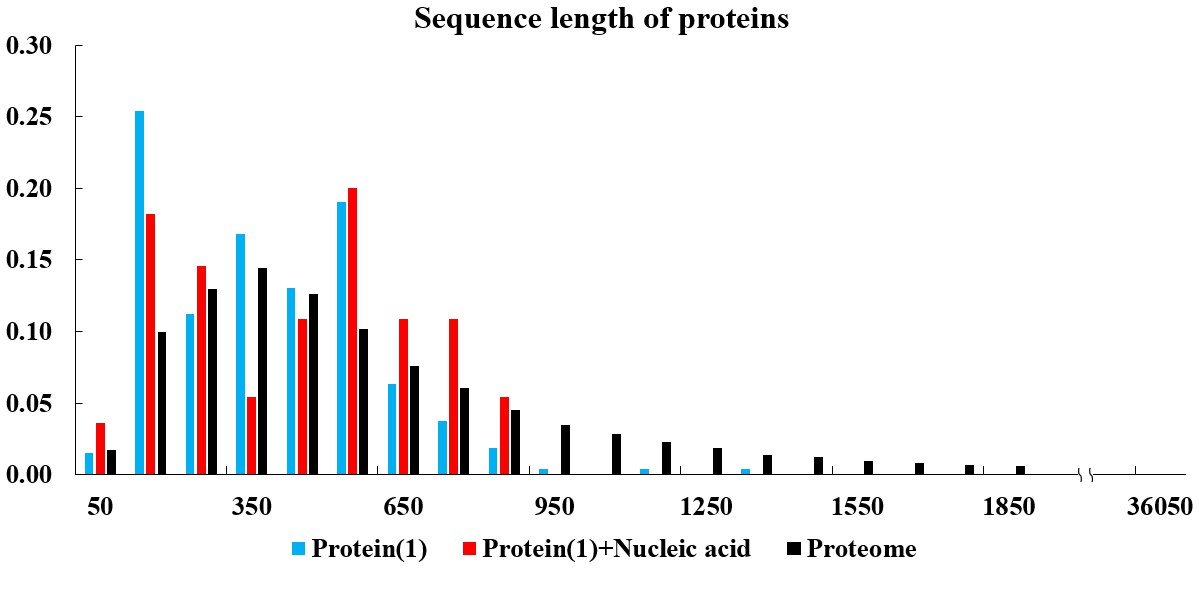
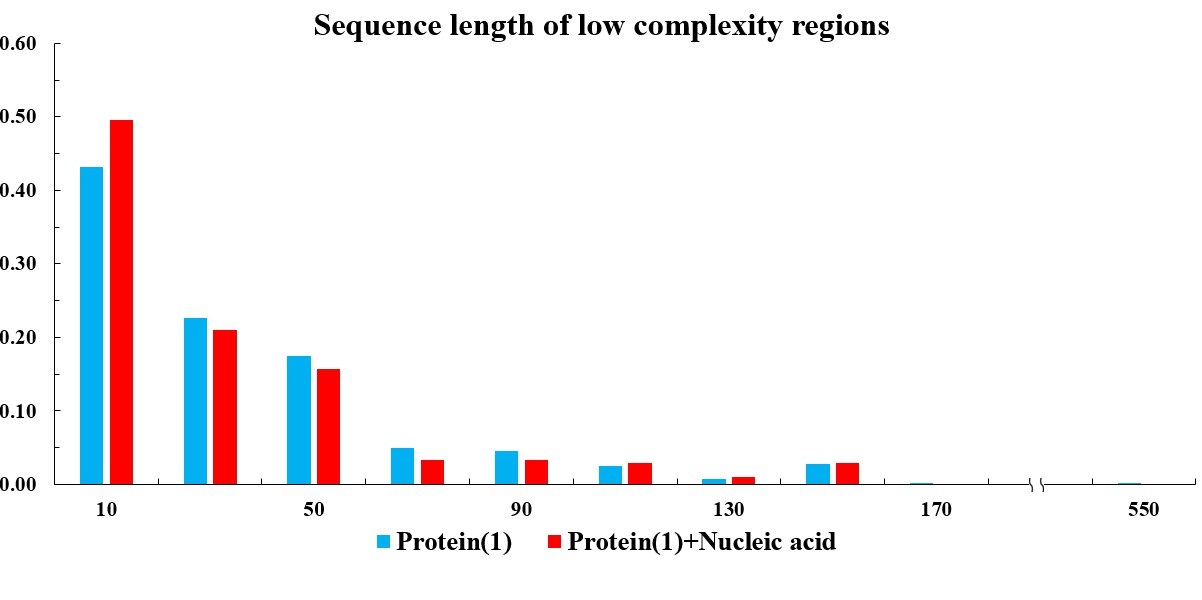
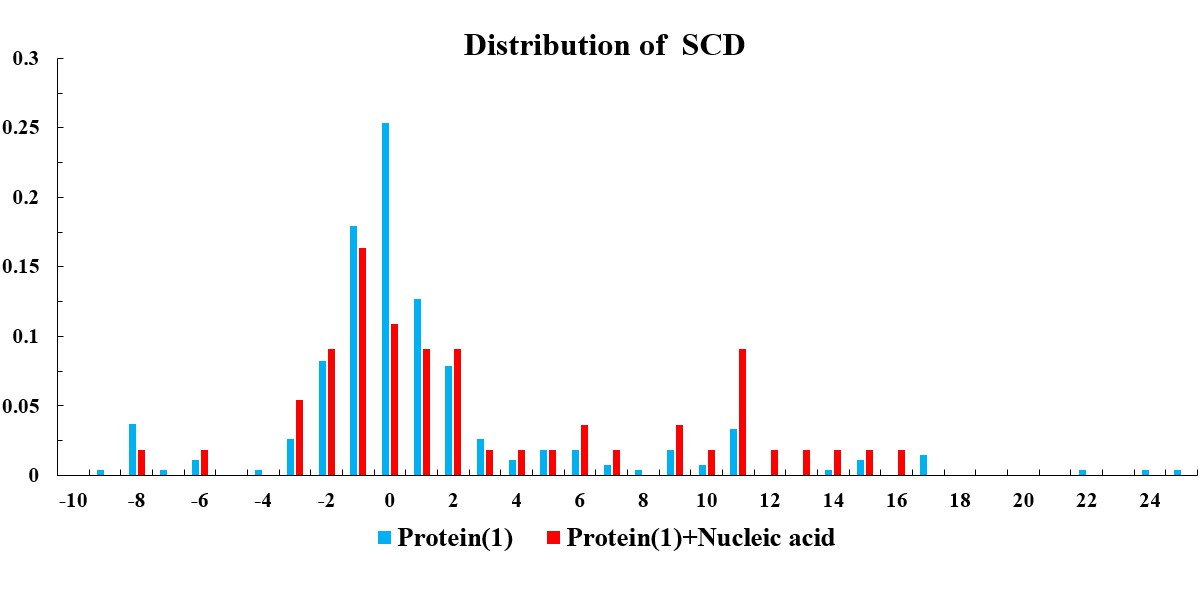
- 1
- 2
- 3
- 4
- 5
- 6
- 7
References
Das, R.K., and Pappu, R.V. (2013). Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc Natl Acad Sci U S A 110, 13392-13397.
Obradovic, Z., Peng, K., Vucetic, S., Radivojac, P., Brown, C.J., and Dunker, A.K. (2003). Predicting intrinsic disorder from amino acid sequence. Proteins 53 Suppl 6, 566-572.
Piovesan, D., Tabaro, F., Paladin, L., Necci, M., Micetic, I., Camilloni, C., Davey, N., Dosztanyi, Z., Meszaros, B., Monzon, A.M., et al. (2018). MobiDB 3.0: more annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res 46, D471-d476.
Sawle, L., and Ghosh, K. (2015). A theoretical method to compute sequence dependent configurational properties in charged polymers and proteins. J Chem Phys 143, 085101.
Wootton, J.C. (1994). Non-globular domains in protein sequences: automated segmentation using complexity measures. Computers & chemistry 18, 269-285.